Evo 2:AI 赋予基因组设计新生命,迎接生物学新时代
2025年2月19日,由Arc Institute、英伟达、斯坦福大学、加州大学伯克利分校和加州大学旧金山分校的顶尖科学家联合发布了革命性生物学大模型——Evo 2。该模型不仅通过AI的强大能力深入理解基因组学的复杂性,还突破性地实现了对基因变异的精准预测与基因组序列的生成,标志着生物学研究迈向新的时代。通过结合9.3万亿个DNA碱基对的训练数据和创新的StrippedHyena 2架构,Evo
2025年2月19日,由Arc Institute、英伟达、斯坦福大学、加州大学伯克利分校和加州大学旧金山分校的顶尖科学家联合发布了革命性生物学大模型——Evo 2。该模型不仅通过AI的强大能力深入理解基因组学的复杂性,还突破性地实现了对基因变异的精准预测与基因组序列的生成,标志着生物学研究迈向新的时代。通过结合9.3万亿个DNA碱基对的训练数据和创新的StrippedHyena 2架构,Evo 2能够提供跨物种的基因变异比较,为生命科学领域带来了前所未有的研究潜力和应用前景。
文章链接:Manuscript | Arc Institute
 图片来源于文章
图片来源于文章
研究背景
生物复杂性与基因组学:所有生命形式都通过 DNA 编码信息。尽管基因组测序、合成和编辑技术已经极大地推动了生物学研究,但要智能地设计新的生物系统,还需要对基因组编码的复杂性有深刻的理解。
人工智能在生物学中的应用:近年来,人工智能(AI)的进步为从大规模数据中发现复杂模式提供了新的框架。Evo 2 是一个基于 AI 的生物基础模型,它通过学习来自所有生命领域的基因组数据来预测基因变异的功能影响,并生成新的基因组序列。
Evo 2 模型介绍
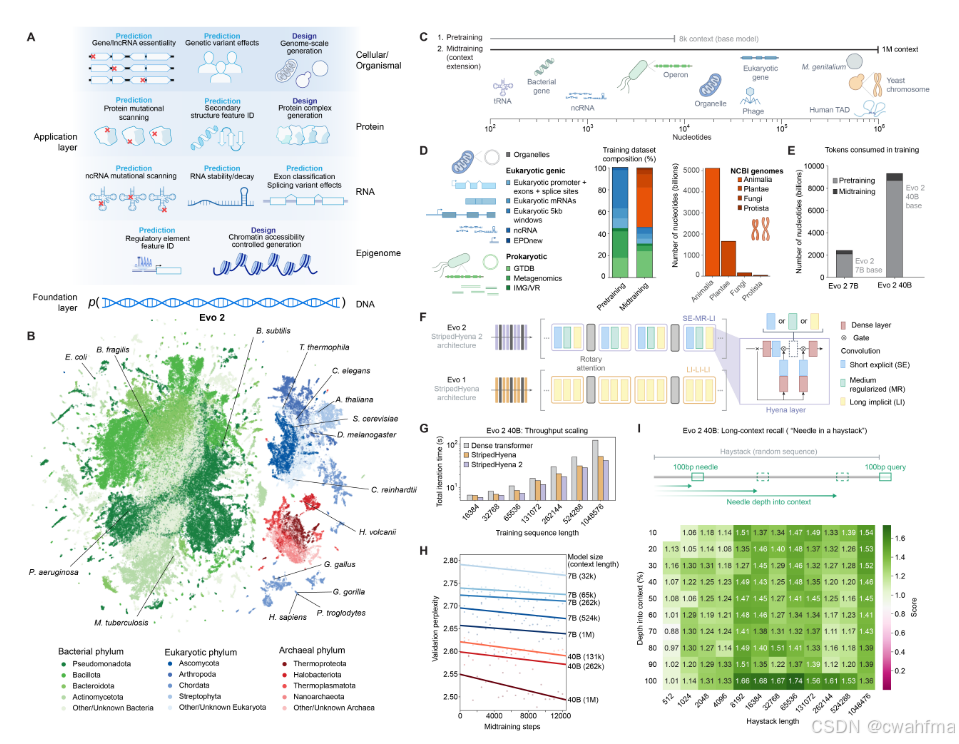
数据集:Evo 2 使用了一个高度策划的基因组图谱进行训练,包含来自细菌、古菌、真核生物和噬菌体的 9.3 万亿个 DNA 碱基对。
模型规模:Evo 2 有两个版本,分别有 70 亿和 400 亿参数,能够处理长达 100 万个碱基对的上下文窗口。
架构:Evo 2 使用了 StripedHyena 2 架构,这是一种新的卷积混合架构,结合了输入依赖的卷积和注意力机制,提高了训练效率和性能。
Evo 2 的功能
预测基因变异的影响:Evo 2 能够准确预测基因变异对蛋白质功能、RNA 功能和生物体适应性的影响,无需针对特定任务进行微调。在预测非编码变异的致病性方面,Evo 2 达到了新的高度,尤其是在 BRCA1 基因变异的分类中表现出色。Evo 2 还能够预测人类临床变异的致病性,包括编码和非编码区域的变异。
基因组序列生成:Evo 2 能够生成线粒体、原核生物和真核生物的基因组规模序列,比以往的方法更具自然性和连贯性。通过在推理时进行搜索,Evo 2 可以实现可控的表观基因组结构生成,例如设计具有特定染色质可及性模式的 DNA 序列。
模型的可解释性:Evo 2 的内部表示能够捕捉多种生物特征,包括外显子-内含子边界、转录因子结合位点、蛋白质结构元素和噬菌体基因组区域。
通过稀疏自编码器(SAE),研究人员能够从 Evo 2 中提取与生物功能相关的特征,例如识别噬菌体区域、移动遗传元件和蛋白质二级结构。
Evo 2 模型在预测基因变异方面具体表现
Evo 2 模型在预测基因变异方面表现出色,尤其是在处理非编码变异和复杂变异类型时,达到了新的高度。以下是其在不同变异类型和数据集上的具体表现:
1. 预测编码和非编码变异的致病性
ClinVar 数据集:Evo 2 在 ClinVar 数据集上对编码和非编码变异的致病性进行了零样本预测(zero-shot prediction)。ClinVar 是一个包含人类疾病相关变异注释的数据库,涵盖了编码和非编码区域的单核苷酸变异(SNV)和非 SNV 类型。
编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.841 和 0.830,虽然略低于 AlphaMissense(0.958)和 ESM-1b(0.923),但仍然表现出色。
非编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.987 和 0.977,显著优于其他模型,包括 PhyloP(0.977)和 GPN-MSA(0.981)。
非编码非 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.971 和 0.934,再次显著优于其他模型,如 PhyloP(0.926)和 Evo 1(0.530)。
综合表现:在编码和非编码变异的综合评估中,Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.970 和 0.960,表现出强大的综合预测能力。
2. 预测剪接变异(SpliceVarDB)
SpliceVarDB 数据集:该数据库包含经过实验验证的剪接变异数据,用于评估模型对剪接相关变异的预测能力。
外显子 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.684 和 0.681,接近 GPN-MSA(0.675)。
内含子 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.926 和 0.930,显著优于其他模型,如 PhyloP(0.898)和 GPN-MSA(0.900)。
综合表现:在内外显子 SNV 的综合评估中,Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.815 和 0.825,表现出色。
3. BRCA1 和 BRCA2 基因变异
BRCA1 数据集:Evo 2 在 BRCA1 基因变异的预测中表现出色,尤其是在非编码变异的预测上。
编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.843 和 0.823,接近 AlphaMissense(0.889)。
非编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.974 和 0.959,显著优于其他模型,如 PhyloP(0.898)和 GPN-MSA(0.918)。
综合表现:在编码和非编码变异的综合评估中,Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.901 和 0.904,表现出强大的综合预测能力。
BRCA2 数据集:Evo 2 在 BRCA2 基因变异的预测中也表现出色。
编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.761 和 0.713,接近 GPN-MSA(0.794)。
非编码 SNV:Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.899 和 0.864,显著优于其他模型,如 PhyloP(0.870)和 GPN-MSA(0.900)。
综合表现:在编码和非编码变异的综合评估中,Evo 2 40B 和 7B 模型的 AUROC 分别达到了 0.842 和 0.828,表现出色。
4. 零样本预测(Zero-shot Prediction)
Evo 2 通过零样本预测展示了其强大的泛化能力。它能够在没有针对特定任务进行微调的情况下,直接利用模型的内部表示来预测变异的功能影响。
在多个基准测试中,Evo 2 的零样本预测性能显著优于其他模型,尤其是在非编码变异和复杂变异类型上。
5. 监督学习分类器
除了零样本预测,Evo 2 的嵌入(embeddings)还可以用于训练监督学习分类器,进一步提升预测性能。
例如,在 BRCA1 基因变异的分类中,基于 Evo 2 嵌入的监督学习分类器达到了 0.94 的 AUROC,显著优于其他基准模型。
Evo 2 模型应用于其他类型的基因变异预测
Evo 2 模型不仅在常见的基因变异类型(如单核苷酸变异 [SNV] 和小片段插入/缺失 [indels])上表现出色,还具备广泛的潜力应用于其他类型的基因变异预测。以下是 Evo 2 在不同类型基因变异预测中的应用和表现:
1. 非编码变异(Non-coding Variants)
非编码变异是指发生在基因组非编码区域的变异,这些区域虽然不直接编码蛋白质,但在基因调控、转录调控和表观遗传调控中起着重要作用。Evo 2 在非编码变异的预测上表现尤为突出:
ClinVar 数据集:Evo 2 在非编码 SNV 和非 SNV 类型的变异上达到了极高的 AUROC 分数(非编码 SNV:0.987,非编码非 SNV:0.971),显著优于其他模型,如 PhyloP 和 GPN-MSA。
SpliceVarDB 数据集:Evo 2 在内含子区域的变异预测上也表现出色,AUROC 分数达到 0.926,接近 GPN-MSA(0.900)和显著优于 PhyloP(0.898)。
BRCA1/BRCA2 数据集:Evo 2 在 BRCA1 和 BRCA2 基因的非编码变异预测上也表现出色,AUROC 分数分别达到 0.974 和 0.899,显著优于其他模型。
2. 剪接变异(Splicing Variants)
剪接变异是指影响 RNA 剪接过程的变异,通常发生在内含子和外显子的交界区域。Evo 2 在剪接变异的预测上也表现出色:
SpliceVarDB 数据集:Evo 2 在内含子和外显子区域的剪接变异预测上均表现出色,外显子区域的 AUROC 分数达到 0.684,内含子区域的 AUROC 分数达到 0.926,接近或优于其他模型。
3. 复杂变异(Complex Variants)
复杂变异包括大片段插入/缺失、结构变异(如倒位、易位)和多核苷酸变异。Evo 2 的长序列建模能力和单核苷酸分辨率使其能够处理这些复杂变异:
长序列建模:Evo 2 的上下文窗口长达 100 万个碱基对,使其能够捕捉长距离的基因组结构和调控关系,这对于复杂变异的预测非常关键。
零样本预测:Evo 2 通过零样本预测展示了其对复杂变异的泛化能力,尤其是在非编码区域的长距离调控元件和结构变异的预测上。
4. 表观遗传变异(Epigenetic Variants)
表观遗传变异是指影响基因表达而不改变 DNA 序列的变异,如染色质可及性、DNA 甲基化和组蛋白修饰。Evo 2 可以通过与表观遗传预测模型(如 Enformer 和 Borzoi)结合,实现对表观遗传变异的预测:
染色质可及性设计:Evo 2 可以通过推理时搜索生成具有特定染色质可及性模式的 DNA 序列,这为研究表观遗传调控提供了新的工具。
5. 其他潜在应用
调控元件变异:Evo 2 可以用于预测影响增强子、启动子和其他调控元件的变异。
长非编码 RNA(lncRNA)变异:Evo 2 在 lncRNA 的功能预测和变异影响评估方面也表现出色,尤其是在预测 lncRNA 本质性(essentiality)方面。
跨物种变异比较:Evo 2 的多物种训练数据使其能够进行跨物种的变异比较和进化分析。
Evo 2 支持跨物种变异比较
Evo 2 支持跨物种变异比较,并且这是其设计和训练的一个重要特点。Evo 2 的训练数据涵盖了来自所有生命领域的基因组序列,包括细菌、古菌、真核生物和噬菌体,这使得它能够学习到跨物种的通用基因组模式和进化规律。以下是 Evo 2 在跨物种变异比较方面的一些关键特点和表现:
1. 多物种训练数据
Evo 2 的训练数据集(OpenGenome2)包含来自不同生物领域的基因组序列,具体如下:
细菌和古菌:超过 113,000 个代表性基因组。
真核生物:包括动物、植物、真菌和原生生物的基因组。
噬菌体和质粒:来自不同宿主的噬菌体和质粒序列。
这种多物种的数据组合使得 Evo 2 能够学习到不同生物之间的进化关系和保守性,从而支持跨物种的变异比较。
2. 进化信号的学习
Evo 2 的内部表示能够捕捉到进化信号,这使得它能够识别不同物种之间的保守区域和功能元件。例如:
Evo 2 学习到了不同物种中保守的外显子-内含子边界、转录因子结合位点和蛋白质结构域。
Evo 2 还能够识别移动遗传元件(如噬菌体和 CRISPR 阵列)在不同物种中的分布。
3. 跨物种变异比较的具体应用
Evo 2 可以用于以下跨物种变异比较的场景:
3.1. 保守性分析
Evo 2 可以评估不同物种中特定基因或基因区域的保守性。例如,通过比较不同物种中相同基因的变异影响,可以识别出在进化上高度保守的区域,这些区域可能具有重要的功能。
3.2. 功能元件的跨物种比较
Evo 2 可以识别不同物种中相似的功能元件,如启动子、增强子和剪接位点。通过比较这些元件在不同物种中的变异影响,可以更好地理解它们的功能和进化动态。
3.3. 病原体与宿主的相互作用
Evo 2 可以用于研究病原体(如噬菌体)与宿主之间的相互作用。例如,通过比较噬菌体感染不同宿主的变异影响,可以识别出宿主基因组中的抗性位点和病原体基因组中的适应性位点。
3.4. 人类变异的进化背景
Evo 2 可以将人类变异放在更广泛的进化背景下进行分析。例如,通过比较人类变异与进化上保守的非编码区域的变异影响,可以更好地评估这些变异的潜在致病性。

火山引擎开发者社区是火山引擎打造的AI技术生态平台,聚焦Agent与大模型开发,提供豆包系列模型(图像/视频/视觉)、智能分析与会话工具,并配套评测集、动手实验室及行业案例库。社区通过技术沙龙、挑战赛等活动促进开发者成长,新用户可领50万Tokens权益,助力构建智能应用。
更多推荐
 47
47 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)