读文献:PepNet:使用预训练的蛋白质语言模型进行抗炎和抗菌肽预测的可解释神经网络
每个圆点代表一个氨基酸残基(例如 I、K、Q…)。序列长度记作 nnn(论文里统一截/填充为 L=40L=40L=40)。把原始序列送入预训练蛋白语言模型(ProtT5-XL-U50)。输出每个残基的,图中标为 n×1024n \times 1024n×1024(每残基 1024 维)。目的是捕获残基在序列中的语义与全局上下文信息(类似 NLP 的词向量)。:每残基的 20 维独热表示,记 n×2
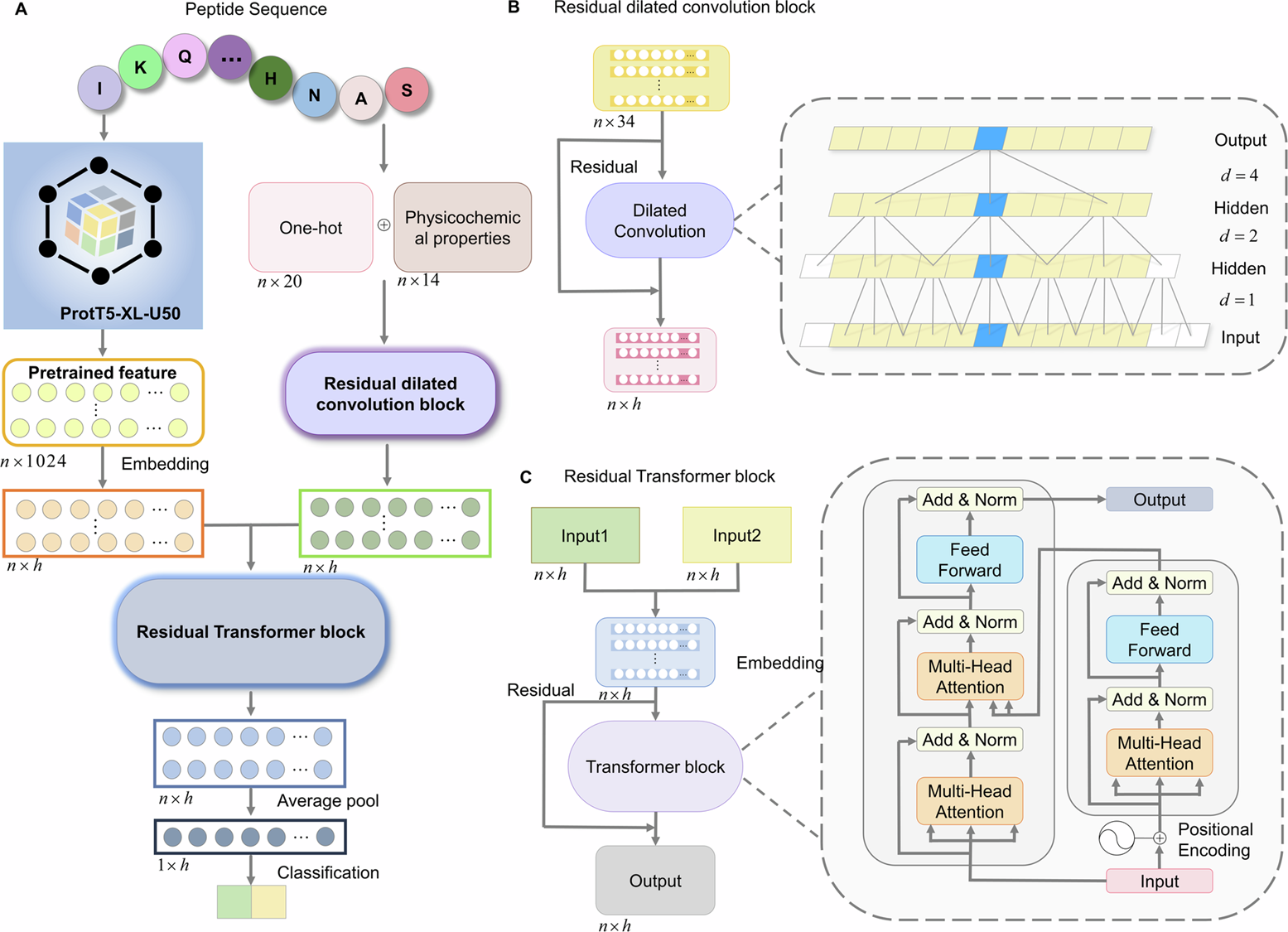
总体流程图详解:图 A 是模型的整体流水线,从肽序列到预测标签的端到端流程:
-
输入 — Peptide Sequence(最上方彩色圆点)
-
每个圆点代表一个氨基酸残基(例如 I、K、Q…)。
-
序列长度记作 nnn(论文里统一截/填充为 L=40L=40L=40)。
-
-
预训练特征(左侧方块,ProtT5-XL-U50) — 黄色矩形
-
把原始序列送入预训练蛋白语言模型(ProtT5-XL-U50)。
-
输出每个残基的 上下文嵌入(embedding),图中标为 n×1024n \times 1024n×1024(每残基 1024 维)。
-
目的是捕获残基在序列中的语义与全局上下文信息(类似 NLP 的词向量)。
-
-
手工/基础特征(中间上部,粉/褐色框)
-
one-hot(20维):每残基的 20 维独热表示,记 n×20n \times 20n×20。
-
physicochemical properties(14维):每残基 14 个理化量度,记 n×14n \times 14n×14。
-
两者拼接后给出基础特征矩阵,合计 n×34n \times 34n×34。
-
-
特征融合(把两类特征合并)
-
预训练 embedding(n×1024n \times 1024n×1024)与基础特征(n×34n \times 34n×34)合并/映射到同一维度 hhh(图中用橙色/绿色小圆块表示 n×hn \times hn×h)。
-
合并后输入后续网络:左路(黄色→橙)和中路(粉/褐→绿)都变为 n×hn \times hn×h。
-
-
Residual Dilated Convolution Block(紫色大框)
-
中间流程先经过残差膨胀卷积模块,用来提取局部与中程模式(后面 B 详细解释)。
-
该模块的输出也是 n×hn \times hn×h。
-
-
Residual Transformer Block(蓝色大框)
-
将卷积得到的局部表示(或融合后的表示)送入残差 Transformer 模块,提取全局依赖(后面 C 详解)。
-
Transformer 的输出仍是 n×hn \times hn×h。
-
-
Average Pool(平均池化)
-
对 n×hn \times hn×h 的序列表示做全局平均池化,得到固定长度向量 1×h1 \times h1×h(不随序列长度变化),便于分类器处理。
-
-
Classification(分类器)
-
平均池化后通过若干全连接层(MLP)输出二分类概率(抗炎 / 非抗炎)。
-
ProtT5 是这篇论文的核心“语义特征来源”
ProtT5 是一个基于 Transformer 的蛋白质语言模型(Protein Language Model),
由 Rostlab 团队(Heinzinger et al., 2021) 开发,用来“理解”氨基酸序列的语义结构。
传统 one-hot 或理化特征只看单个残基,无法理解上下文。(A图的one-hot)
(注意:ProtT5-XL-U50 是个很大的模型(约 3 B 参数),推荐使用 GPU ≥ 16 GB 显存)
这里和前面复现的师哥的DeepBP中运用的ESM-2功能一样(能产出每个残基的向量表示(embedding))
One-hot 编码(独热编码)
把每个氨基酸看成一个离散的符号(类似单词),用一串 0/1 组成的固定长度向量来表示。
| 氨基酸 | One-hot 向量示例 |
|---|---|
| A | [1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0] |
| R | [0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0] |
| N | [0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0] |
| … | … |
缺点:不能体现上下文语义。举例:D(天冬氨酸)与E(谷氨酸)都是酸性残基,但在 one-hot 中完全不相似,这正是其局限。
理化特征(Physicochemical Properties)
例如序列 “ACD”,
[[ 1.8, 8.1, 60, 0, 6.0, ...], # A
[ 2.5, 5.5, 110, 0, 5.5, ...], # C
[-3.5, 13.0,125, -1, 2.8, ...]] # D
图 B 把残差膨胀卷积的内部结构放大说明:(整篇论文架构的第一层特征提取模块。)
在不破坏序列长度的前提下,捕获短程、中程的局部氨基酸模式(motif),并用残差结构保证梯度稳定与特征融合。
也就是说,它负责学“哪些局部氨基酸组合与抗炎功能有关”。
-
输入与输出形状
-
输入:n×34n \times 34n×34(或已映射为 n×hn \times hn×h)——图左上角小黄方块表示输入通道。
-
输出:n×hn \times hn×h(粉色小块)。
-
-
核心单元:Dilated Convolution(膨胀卷积)
-
膨胀卷积(dilated conv)在卷积核之间插入“空洞(dilation)”,以扩大感受野而不增加参数/层数。
-
图右侧小放大图展示了多层膨胀、不同 dilation rates:
-
第 1 层:膨胀率 d=1d=1d=1,看到邻近残基(3-gram 模式);
-
第 2 层:膨胀率 d=2d=2d=2,跳过一个残基,看到更广上下文;
-
第 3 层:膨胀率 d=4d=4d=4,感受野翻倍,能看到更远的残基关联。
每层卷积后都有ReLU + 归一化(LayerNorm/BatchNorm)。
-
-
这样堆叠后,单个输出单元能感知到较长的局部窗口(有效感受野扩大)。
-
-
残差连接(Residual)
-
输入被加回到卷积输出(跳跃连接),形成残差结构(图中灰色箭头回路)。
-
作用:缓解深度网络训练中梯度消失、加速收敛并保留原始信息。
-
-
层归一化/激活
-
实际实现会在每次卷积后加激活(ReLU)和归一化(LayerNorm 或 BatchNorm),保证训练稳定(图中未标出但常见)。
-
-
为什么用它?
-
卷积擅长局部模式(短序列 motif),膨胀可以让卷积看到更宽的上下文但仍保留位置敏感性(顺序信息)。
-
与 Transformer 的全局注意力互补:卷积负责“局部结构化信息”,Transformer 负责“全局关系”。
-
相当于在卷积核内部插入空洞,跳着取输入:
-
普通 kernel=3 → 看相邻 [A, R, N]
-
dilation=2 → 看 [A, N, C](跳过一个)
-
dilation=4 → 看 [A, D, Q](更远)
这让 kernel 在不增加参数的前提下,感受到更长距离的依赖关系。
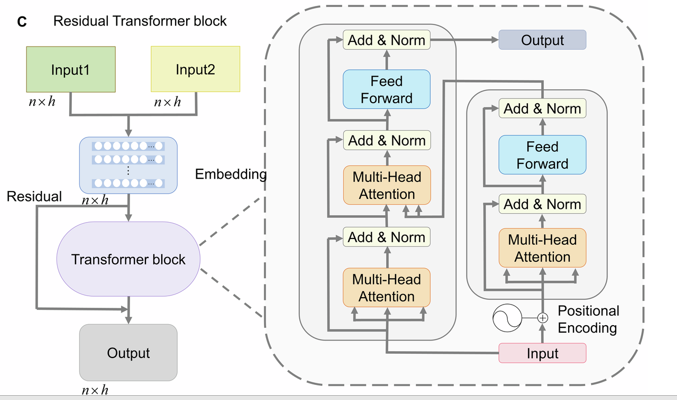
图 C 展示 Transformer 模块和其与残差结构的结合方式:
-
模块输入
-
图中标示有两个输入
Input1和Input2(均 n×hn \times hn×h)。 -
这表示 Transformer 块可以接受来自不同来源的表示(例如预训练 embedding 路径 + RDC 路径),论文实现里通常会拼接或相加这些表示后送入 Transformer(实际细节在代码里)。
-
-
Transformer block(中间蓝色)
-
核心由 Multi-Head Attention(多头注意力) + Feed-Forward Network(前馈网络) 组成。
-
每个子层外面包裹 Add & Norm(残差相加 + 层归一化)。
-
图右侧大放大图展示了标准 Transformer 的两个堆叠子模块(可能是 encoder–decoder 或 双层 encoder):
-
自注意力层:输入经位置信息(Positional Encoding)后计算 Q/K/V 并做多头注意力;
-
前馈层:逐位置的两层 FC(通常是线性→激活→线性);
-
每个子层都有残差连接(输出 = LayerNorm(x + Sublayer(x)))。
-
-
-
残差 + 输出
-
Transformer 输出再与输入做残差(图中左侧有 residual 回路),便于信息流动和稳定训练。
-
最终输出形状 n×hn \times hn×h。
-
-
位置信息(Positional Encoding)
-
Transformer 无法天然感知序列顺序,图中右下角标注了 Positional Encoding(正弦/余弦或可学习的位置编码),加到输入 embedding 上以编码位置信息。
-
-
为什么要 Transformer?
-
自注意力能直接建立任意两残基之间的依赖关系(远程相互作用),适合捕获决定活性的远程协同特征(例如 N 端与 C 端的交互)。
-
Embedding:
假设输入序列为:A R N D C
把每个字母映射成一个 1024维向量(ProtT5输出)
得到矩阵形状 (n=5, h=1024)
那为什么不直接用 1024 维?
因为:
-
ProtT5 的特征虽然强,但维度太高,容易过拟合;
-
后续模型参数量会暴涨(例如 Transformer 的 attention 要计算 h×h 矩阵);
-
不同特征源可能维度不同(ProtT5=1024,理化特征=34),需要统一到同一维度 h。
所以这一层既是“降维层”,也是“特征对齐层”。
Positional Encoding
Positional Encoding 加到 Embedding 上(绝对位置编码)
Transformer 自身不感知序列顺序,
所以要在 embedding 上加上“位置信息”:
X′=X+PositionalEncodingX' = X + \text{PositionalEncoding}X′=X+PositionalEncoding
这样模型既知道“是什么氨基酸”,又知道“它在序列中的位置”
注意力机制(“相似度机制”)
🧩 一、从直觉出发:Q、K、V 的角色
Q、K、V 是三种不同的向量表示,分别表示“我要问的问题”“别人是谁”“别人能提供什么信息”。
| 名称 | 英文 | 含义 | 类比 |
|---|---|---|---|
| Q | Query | “我要找什么样的信息” | 提问 |
| K | Key | “我是什么样的信息” | 值得被查找的关键字 |
| V | Value | “我能提供什么信息” | 被提取的答案内容 |
Attention 的核心是:
每个位置的 Query(Q)去匹配所有位置的 Key(K),
看谁更相关(相似度高),
然后用对应的 Value(V)来加权求和生成新的表示。
🧠 二、举一个实际例子:抗炎肽序列
假设我们有一个短肽序列:A R N 这3 个氨基酸。
模型先经过 embedding 得到输入矩阵 XXX:
假设每个氨基酸是 2 维向量(简化表示):
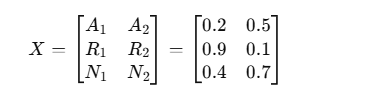
维度是 3×2,其中:
-
3 = 序列长度 (n)
-
2 = embedding 维度 (h)
🧮 三、计算 Q, K, V
Transformer 会把输入 X 乘上三组不同的权重矩阵:(这篇论文使用的是自注意力机制,因为Q、K、V 全来自同一个 X)
![]() (
(
当模型计算完损失(比如预测错误率)后,会进行反向传播:
-
误差梯度从输出层往前传;
-
计算出损失对每个参数的偏导;
-
更新 WQ,WK,WVW_Q, W_K, W_VWQ,WK,WV(以及模型的其他参数)。
所以,这三个矩阵是自动学习到的。
)
假设: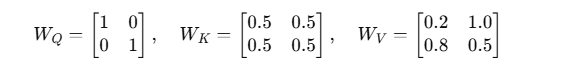
(这里的WQ,WK,WV就是“头”,将原始的Q,K,V投影到另一个空间,最后算出来的O,在✖WO让他投影回去)
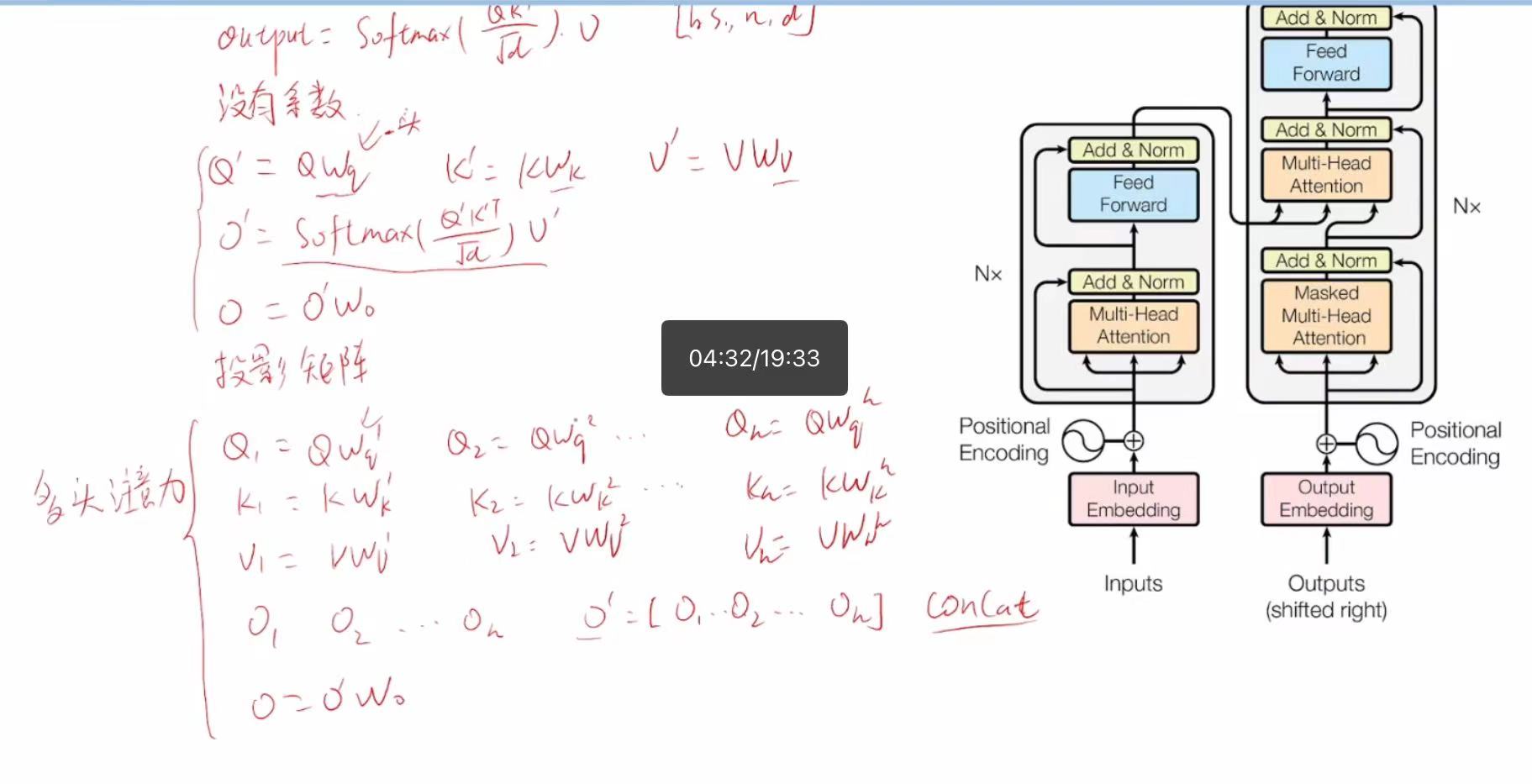
视频链接:【9.多头注意力-哔哩哔哩】 https://b23.tv/b2lojM8
(1) Q = XW_Q
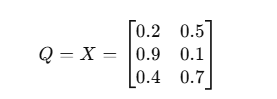
(2) K = XW_K

(3) V = XW_V
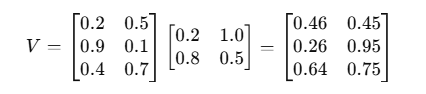
⚙️ 四、计算注意力分数(QKᵀ)

每个元素 (i,j) 表示:第 i 个氨基酸对第 j 个的相关度。
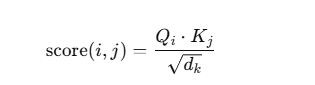
d是一个标量,防止QK相乘后不会大的离谱
🧮 五、softmax 归一化(按行)
以第一个 Query 为例:
![]()
表示:
第 1 个残基 A 关注 R 的概率 0.33,关注 N 的概率 0.37。
对所有 Query 都做同样的 softmax,得到注意力权重矩阵 α:
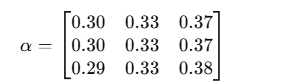
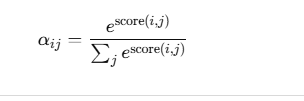
🧮 六、计算输出 O = αV
以第一行举例:
![]()
即第一个残基更新后的表示是:
加权融合了所有残基的 Value(信息),
权重由“关注程度” α 决定。
结果是一个新的矩阵 O(3×2),每个位置的表示都整合了整个序列的上下文。
Transformer 的多头注意力会让每个残基的 embedding 更新成包含全局信息的表示:
“我现在不仅知道自己是谁(Ala),
还知道我左边是 Lys,右边是 Asp,它们都可能影响我。”
于是输出仍是 n×hn \times hn×h,但每个向量都更“聪明”了。
问:为啥这里的Input分成了两路,一路没有位置编码,一路有位置编码,然后再其中一个多头注意力汇合了?
🔍 直观理解(以抗炎肽为例)
假设肽序列是:K Q H N A S
-
有些特征取决于序列顺序,比如:
“K” 靠近 “Q” 可能影响氢键或构象;
“K” 和 “H” 的相对位置可能决定电荷分布;
→ 这些就依赖 位置编码的注意力路径。 -
另一些特征是全局的,比如:
“K” 和 “S” 虽然相隔远,但共同决定抗炎功能;
→ 这些由 不带位置编码的注意力路径 捕获。
最后两路输出在上层汇合,让模型既懂顺序局部性,又懂全局协同性。
Average Pooling 过程
假设:K Q H N A S(这条肽) n=6,h=4
| 残基 | Transformer 输出特征向量(4维) |
|---|---|
| K | [0.8, 0.1, 0.3, 0.6] |
| Q | [0.6, 0.2, 0.5, 0.5] |
| H | [0.5, 0.4, 0.4, 0.4] |
| N | [0.4, 0.5, 0.5, 0.3] |
| A | [0.3, 0.6, 0.4, 0.2] |
| S | [0.2, 0.7, 0.3, 0.1] |
它们代表每个残基在整个序列上下文中“扮演的角色”。
例如:
-
“K” 可能携带强正电信号;
-
“S” 和 “A” 代表疏水或极性残基;
-
模型会自动学习它们的“协同模式”。
平均池化就是对所有残基的表示取平均,得到一个全局序列表示。
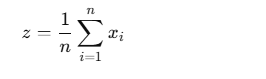
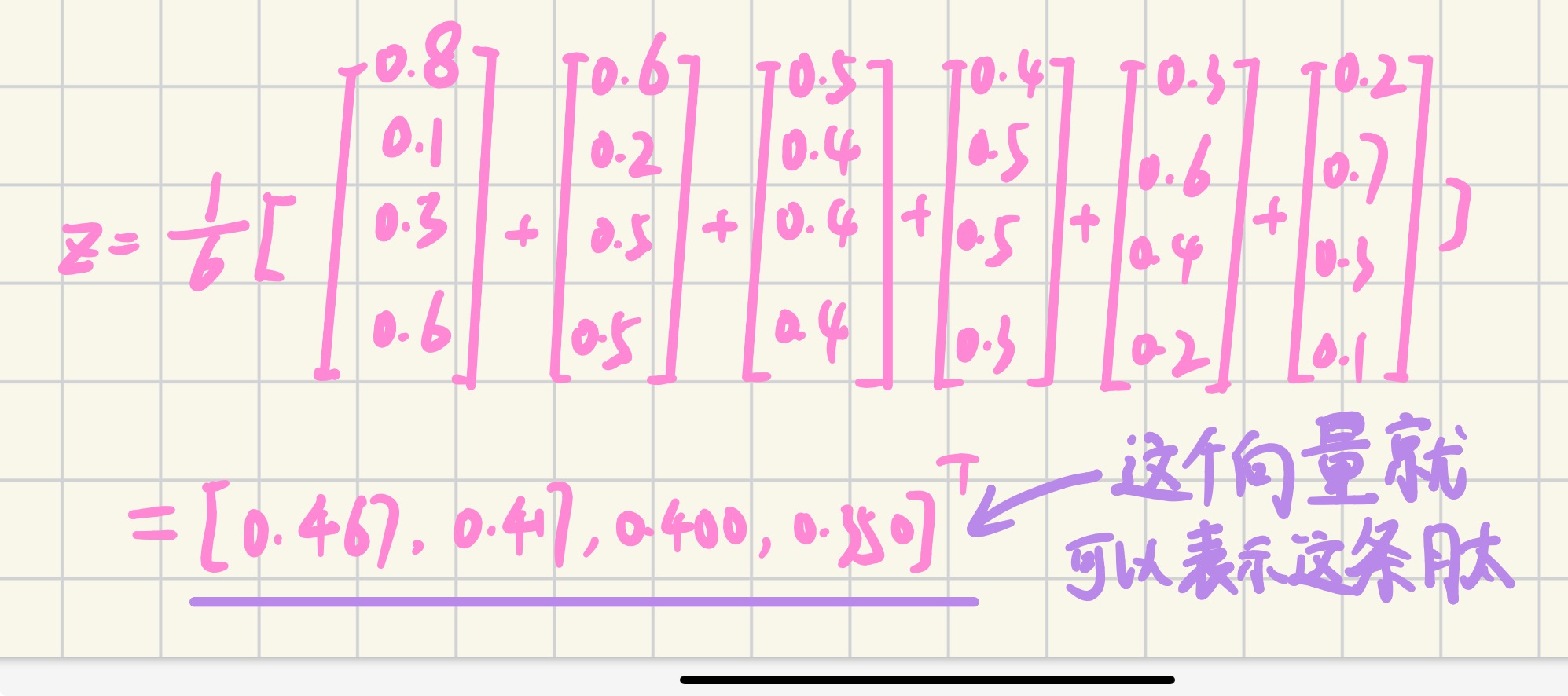
这就是整条肽链的综合表征:
-
第1维可能对应“总体正电特征”
-
第2维可能对应“整体极性”
-
第3、4维可能对应“局部氢键倾向”“柔性”等
这些含义不会直接给出,而是模型自己学到的。
送入分类器(MLP)
全连接层权重示意(隐藏层维度 2):
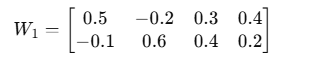
计算:
![]()
得到隐藏特征h=[0.44,0.53]。
再经过最后一层:![]()
例如输出: y=0.87
✅ 这代表模型认为该肽是“抗炎肽”的概率为 87%。

火山引擎开发者社区是火山引擎打造的AI技术生态平台,聚焦Agent与大模型开发,提供豆包系列模型(图像/视频/视觉)、智能分析与会话工具,并配套评测集、动手实验室及行业案例库。社区通过技术沙龙、挑战赛等活动促进开发者成长,新用户可领50万Tokens权益,助力构建智能应用。
更多推荐
 20
20 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)