CEL结尾的芯片数据处理方法(oligo和affy)学习
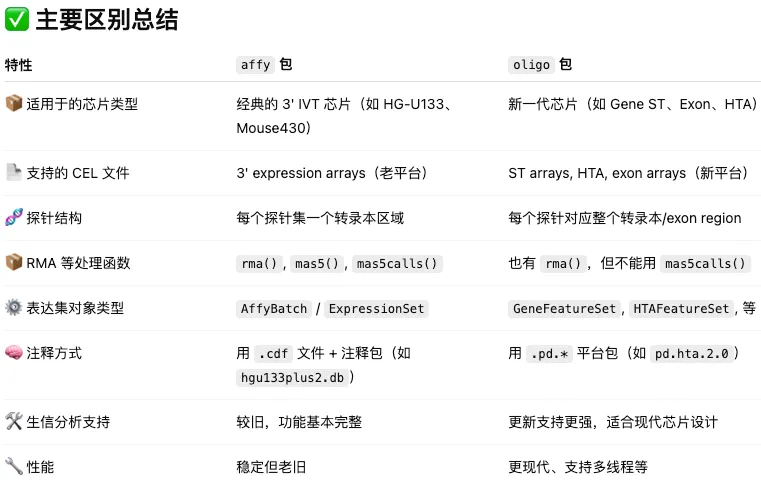
oligo和affy这两个包都能够处理CEL结尾的芯片数据,但是适用性存在一定的差别。让大模型帮忙整理了一下。感觉回答的还挺好。
·
oligo和affy这两个包都能够处理CEL结尾的芯片数据,但是适用性存在一定的差别。让大模型帮忙整理了一下。
感觉回答的还挺好。

oligo包分析流程
选择一个适合oligo包的CEL数据
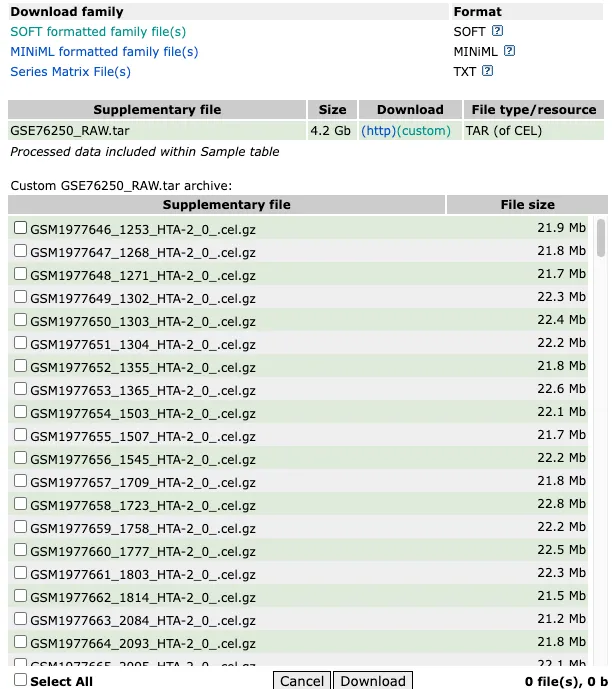
提供一个解压缩并且移动文件的思路代码
# 解压缩
library(R.utils)
# 设置原始目录(包含 .gz 文件)
input_dir <- "~/Desktop/GSE76250/GSE76250_RAW/"
# 设置解压后的目标目录(确保已存在)
output_dir <- "~/Desktop/GSE76250/GSE76250_new/"
dir.create(output_dir, showWarnings = FALSE)
# 获取所有 .gz 文件路径
gz_files <- list.files(input_dir, pattern = "\\.gz$", full.names = TRUE)
# 解压到新的文件夹中
for (f in gz_files) {
gunzip(filename = f,
destname = file.path(output_dir, sub("\\.gz$", "", basename(f))),
overwrite = FALSE,
remove = FALSE) # 设置为 FALSE 不删除原文件
}
正式流程
library(oligo)
# 设置路径
dir_cels='/Users/zaneflying/Desktop/GSE76250/GSE76250_new/'
cel_files <- list.celfiles(dir_cels, full.names = TRUE)
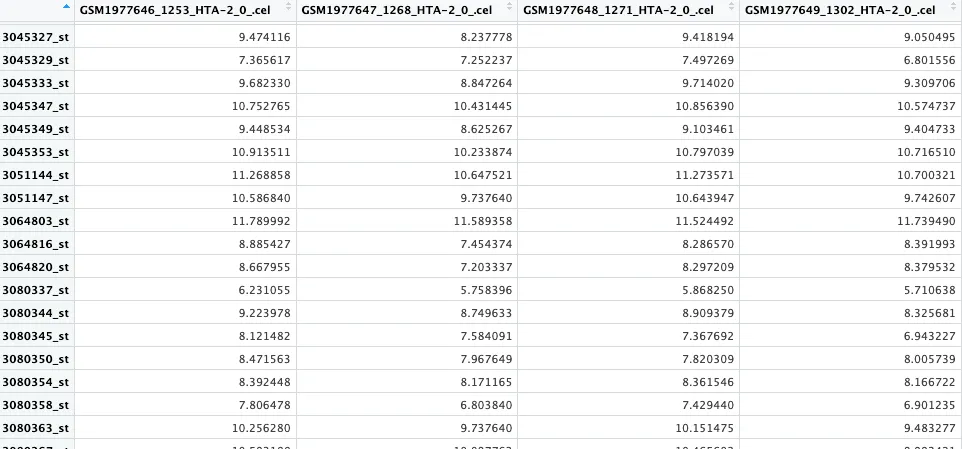
affy_data <- read.celfiles(cel_files)
expr_matrix <- exprs(eset) # 获取表达量数据
affy包分析流程
选择一个适合affy包的CEL数据
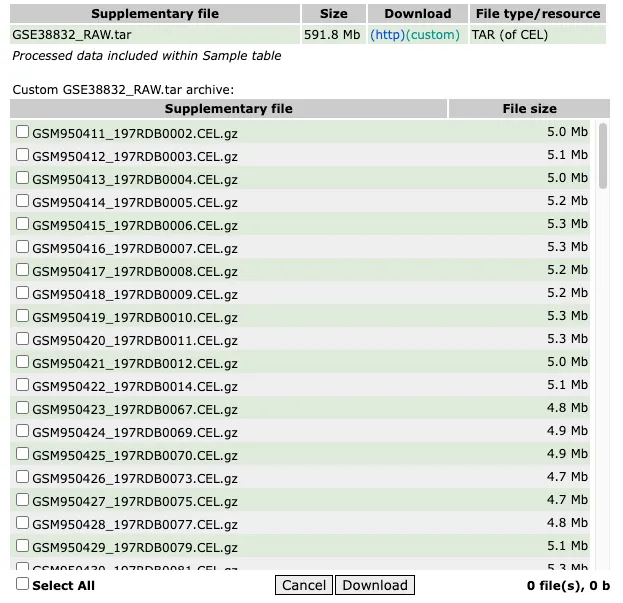
library(affy)
getwd()
dir_cels='~/Desktop/GSE76250/GSE76250_RAW/'
#affy_data = ReadAffy(celfile.path=dir_cels)
eSet = rma(affy_data) #rma函数去normalization
##要进行过滤
calls = mas5calls(affy_data) # get PMA calls
calls = exprs(calls)
absent = rowSums(calls == 'A') # how may samples are each gene 'absent' in all samples
absent = which(absent == ncol(calls)) # which genes are 'absent' in all samples
eSet = eSet[-absent,] # filters out the genes 'absent' in all samples
save(eSet,file = "GSE38832_afterCEL.Rdata")
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟 。

火山引擎开发者社区是火山引擎打造的AI技术生态平台,聚焦Agent与大模型开发,提供豆包系列模型(图像/视频/视觉)、智能分析与会话工具,并配套评测集、动手实验室及行业案例库。社区通过技术沙龙、挑战赛等活动促进开发者成长,新用户可领50万Tokens权益,助力构建智能应用。
更多推荐
 3
3 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)