深入解析生物信息学中的CHIP-Seq数据分析
CHIP-Seq(Chromatin Immunoprecipitation Sequencing)是一种高通量测序技术,它将染色质免疫沉淀(ChIP)与大规模DNA测序相结合,用于识别蛋白质与DNA相互作用的精确位置。自2007年首次提出以来,CHIP-Seq技术迅速发展,并已成为研究基因调控网络的重要工具。峰值检测是指在CHIP-Seq数据分析过程中,识别出基因组上具有显著信号强度的区域。这些

简介:CHIP-Seq技术是研究蛋白质-DNA相互作用的重要工具,尤其在理解基因表达调控方面发挥了巨大作用。本文介绍从实验准备到生物信息学分析的 CHIP-Seq 数据分析流程,包括质量控制、数据预处理、读取对齐、峰值检测、峰注释、差异分析、功能富集、网络构建以及结果可视化等关键步骤。 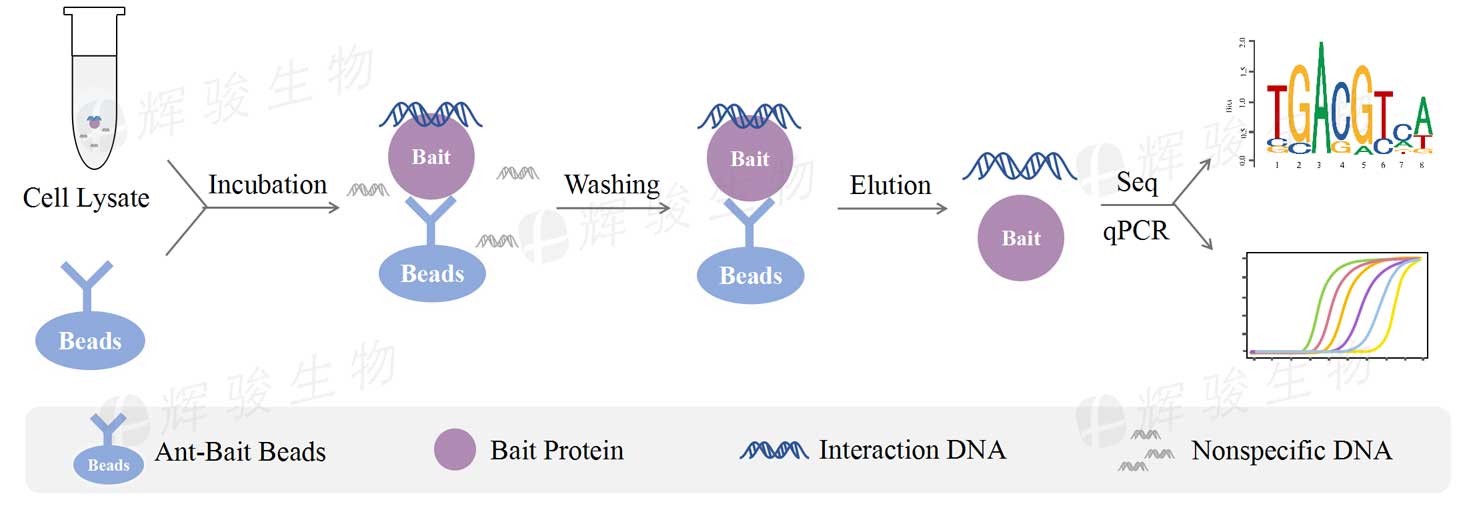
1. CHIP-Seq 技术概览
CHIP-Seq技术的定义与发展历程
CHIP-Seq(Chromatin Immunoprecipitation Sequencing)是一种高通量测序技术,它将染色质免疫沉淀(ChIP)与大规模DNA测序相结合,用于识别蛋白质与DNA相互作用的精确位置。自2007年首次提出以来,CHIP-Seq技术迅速发展,并已成为研究基因调控网络的重要工具。
CHIP-Seq技术的核心原理
CHIP-Seq技术的核心原理包括以下步骤:首先通过抗体特异性结合目标蛋白质并沉淀染色质片段;接着,这些片段经过纯化、打断,并与接头连接进行测序;最后,测序得到的短序列读段(reads)与参考基因组进行比对,从而确定蛋白质结合的位置。通过这一系列的实验和计算方法,研究人员可以准确地识别基因组上特定蛋白质的结合位点。
CHIP-Seq与其他基因组分析技术的比较
CHIP-Seq与传统的ChIP-chip技术相比,其主要优势在于更高的分辨率和更宽的动态范围。与全基因组关联研究(GWAS)相比,CHIP-Seq更能精确地映射出蛋白质与DNA的相互作用位点。此外,CHIP-Seq与RNA-Seq技术一样,都需要进行高质量的序列数据生成和处理。然而,CHIP-Seq专注于基因调控区域的检测,而RNA-Seq则侧重于基因表达水平的分析。
2. 质量控制过程与工具
2.1 CHIP-Seq数据质量控制的重要性
2.1.1 质量控制的基本流程
质量控制(Quality Control, QC)是 CHIP-Seq 数据分析的关键步骤。它确保了数据分析的有效性和可靠性。基本流程涉及原始数据的检查、数据清洗、对齐质量检查、重复率分析和标准化处理。
原始数据检查 通常包括对数据文件格式的确认和数据质量的初步评估,如查看序列的长度分布和质量分数。在 CHIP-Seq 分析中,原始数据通常来源于 Illumina 或其他高通量测序平台。
数据清洗 涉及到去除低质量的序列、修剪接头序列和去除污染序列等。低质量序列可能含有测序错误导致的插入、删除或不准确的碱基。高质量序列是后续分析可靠性的基础。
对齐质量检查 包括检查序列在参考基因组上的比对效率、是否存在大量的不一致对齐等。比对的准确性对于峰的准确检测至关重要。
重复率分析 是指检查比对到基因组相同位置的重复序列。这种分析有助于识别 PCR 扩增导致的错误信号。
标准化处理 是调整不同样本之间数据差异的过程,保证了数据分析的公平性和可比较性。
2.1.2 常用的质量控制工具与软件
在 CHIP-Seq 数据分析中,有许多高效的工具可用于质量控制。其中一些广泛使用的包括:
- FastQC :用于检测原始测序数据的质量,并提供报告。
- TrimGalore :用于去除低质量的序列和测序接头。
- Picard 和 SAMtools :用于分析比对到参考基因组后的 BAM 文件质量,提供各种统计信息。
- bedtools :用于分析和处理基因组学数据集。
2.1.3 质量控制中的关键参数解释
在进行质量控制时,需要对多个参数进行解读和解释。比如:
- 质量分数(Quality Score) :通常表示为 ASCII 码,可以反映测序读段的质量。
- GC 含量 :GC 含量的异常分布可能指示测序过程存在问题。
- 序列重复度(Duplication Rate) :高水平的序列重复度可能表明样本准备过程中的 PCR 扩增偏差。
2.2 CHIP-Seq实验设计的考量因素
2.2.1 选择适当的芯片类型
CHIP-Seq 实验中选择正确的芯片类型对于成功的实验至关重要。芯片类型取决于研究的目的和目标蛋白。例如,用于检测组蛋白修饰的芯片和用于分析转录因子结合的芯片有所不同。
2.2.2 实验材料与条件的标准化
实验材料和条件的标准化可以减少实验误差和提高数据的可重复性。这包括细胞培养的条件、芯片处理的条件和样本处理过程。
2.2.3 预实验与优化策略
预实验是 CHIP-Seq 实验设计的一个重要组成部分。它们用于测试和优化实验条件,以确保主实验的成功。
2.3 质量控制中的常见问题及解决方案
2.3.1 噪声和背景的识别与处理
噪声可能来源于技术原因或生物学背景。可以采用不同的方法,如移除低复杂度的序列和控制输入 DNA 的量来减少噪声和背景。
2.3.2 片段大小分布的评估
在 CHIP-Seq 分析中,片段大小分布对于峰值检测非常关键。理想的片段大小分布会表现出明显的模式,如单峰或双峰分布。
2.3.3 样本间变异的检测与校正
样本间变异可能是由于实验技术或生物学原因。使用适当的统计方法,例如标准化因子校正,可以减少这些变异带来的影响。
在下一章节中,我们将深入探讨数据预处理方法,包括数据清洗、读段对齐以及数据标准化与归一化的技术。这一步骤对于后续分析至关重要,能够确保数据的质量和准确性。
3. 数据预处理方法
在基因组学研究中,高质量的数据预处理是获取可信赖分析结果的基石。 CHIP-Seq 数据预处理方法包括数据清洗、读段对齐的前处理以及数据标准化与归一化等多个环节,每个环节都至关重要。
3.1 数据清洗的重要性
3.1.1 去除污染和无关序列
CHIP-Seq 数据通常来自高通量测序平台,这些数据中除了目标 DNA 片段外,还可能夹杂着由测序过程中产生的污染序列、PCR 引物和接头序列等。首先必须清理掉这些无关序列,以避免其影响后续的数据分析。
操作步骤:
- 使用
cutadapt或trimmomatic工具去除接头序列。 - 利用
fastq_screen检测污染情况并过滤掉污染序列。
代码示例:
# 使用 cutadapt 去除接头序列
cutadapt -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -o output.fastq input.fastq
# 使用 fastq_screen 清理污染序列
fastq_screen --conf fastq_screen.conf input.fastq
3.1.2 低质量读段的过滤
为了保证数据质量,需要过滤掉质量低下的读段。这一步骤涉及去除那些包含太多错误测序碱基的读段,因为它们可能导致错误的基因组定位和假阳性。
操作步骤:
- 利用
fastp工具进行质量控制(QC)。 - 根据质量评分阈值过滤掉低质量读段。
代码示例:
# 使用 fastp 进行质量过滤
fastp --in1 input_R1.fastq --in2 input_R2.fastq --out1 filtered_R1.fastq --out2 filtered_R2.fastq --qualified_quality_phred 20 --unqualified_percent_limit 50
3.1.3 PCR 重复序列的识别与剔除
在 DNA 片段扩增过程中产生的 PCR 重复序列可能误导峰值检测,导致错误的生物学推论。因此,需要使用特定的算法识别并剔除这些重复。
操作步骤:
- 使用
MarkDuplicates工具(来自 Picard 包)来标记重复的读段。 - 从分析中移除这些重复的读段。
代码示例:
# 使用 Picard 的 MarkDuplicates 工具识别并标记重复
java -jar picard.jar MarkDuplicates INPUT=dedupped.bam OUTPUT=dedupped_marked.bam METRICS_FILE=metrics.txt REMOVE_DUPLICATES=false
# 移除重复序列后得到最终的 BAM 文件
samtools view -F 0x400 -b dedupped_marked.bam > final_dedupped.bam
3.2 读段对齐的前处理
3.2.1 建立参考基因组索引
在读段对齐前,需要在参考基因组上构建索引文件,这有助于提高对齐的效率和准确性。
操作步骤:
- 使用
bwa、bowtie或hisat2等工具创建索引。 - 选择合适的引物序列和基因组版本。
代码示例:
# 使用 bwa 构建参考基因组索引
bwa index -a bwtsw human_genome.fa
3.2.2 配准和映射策略
配准和映射是将读段对准参考基因组的过程,这一步骤的关键是选择正确的对齐工具和参数。
操作步骤:
- 使用
bwa mem、bowtie2或hisat2等工具将读段映射到参考基因组。 - 验证对齐质量,如对齐率和唯一映射读段的比例。
代码示例:
# 使用 bwa mem 将读段对齐到基因组
bwa mem human_genome.fa input_R1.fastq input_R2.fastq > aligned.bam
3.2.3 多重比对与去重流程
多重比对读段可能导致分析复杂化,通常需要去重,保留最优的比对结果。
操作步骤:
- 使用
samtools markdup对比对后的 BAM 文件去重。 - 分析去重后的数据,确认去重效率和结果的可靠性。
代码示例:
# 使用 samtools 去除多重比对的读段
samtools markdup -r -s input.bam output.bam
3.3 数据标准化与归一化
3.3.1 数据标准化的必要性
为了消除测序深度、样本间差异和实验操作偏差对分析结果的影响,需要对数据进行标准化处理。
3.3.2 不同标准化方法的比较
不同标准化方法适用于不同的分析场景。例如,TPM(Transcripts Per Million)适用于转录组表达分析,而 RPKM(Reads Per Kilobase per Million mapped reads)或 FPKM(Fragments Per Kilobase per Million mapped reads)则适用于基因表达分析。
3.3.3 归一化的技术与实践
标准化后,数据还需要进一步归一化处理,以减少系统偏差和非生物变异的影响。
操作步骤:
- 使用专门的生物信息学软件(如
DESeq2或edgeR)进行归一化。 - 分析归一化前后的数据分布和变化,评估归一化效果。
代码示例:
# 使用 DESeq2 进行归一化处理
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData,
design = ~ sample + condition)
dds <- DESeq(dds)
normalized_counts <- counts(dds, normalized=TRUE)
通过上述步骤,CHIP-Seq 数据预处理的各个环节得以实现,为后续的峰值检测和分析工作打下坚实基础。
4. 高通量测序数据对齐
4.1 测序数据对齐的基本概念
4.1.1 对齐算法的原理
高通量测序数据对齐是指将短的测序读段(reads)与参考基因组进行比对的过程。对齐算法的基本原理是寻找读段与参考基因组之间的最佳匹配位置。这一过程通常涉及以下几个关键步骤:读段的读取,参考基因组的选择,序列比对,以及后续的处理步骤。
为了理解对齐算法的工作原理,首先要认识到基因组序列可以被视作一个长字符串,而测序得到的读段则是这个长字符串中的子串。对齐算法就是用来识别这些子串在长字符串中的准确位置,并处理任何错配、插入或缺失的情况。这类似于在文档中查找一段特定的文字,需要找到精确的起始位置以及所有可能的变异。
对齐算法可以分为两大类:全局对齐与局部对齐。全局对齐尝试对齐整个读段和参考序列的对应部分,而局部对齐则是寻找读段中与参考序列高度相似的较短区域,这在寻找基因组中的重复元素时尤为有用。
4.1.2 常用的测序数据对齐工具
在 CHIP-Seq 实验和高通量测序数据处理中,有多个常用工具可以执行这些对齐任务。一些广泛使用的对齐工具包括 BWA、Bowtie、Bowtie2 和 STAR 等。
- BWA 是一个为 Illumina 测序平台设计的对齐工具,它使用 Burrows-Wheeler 转换算法来实现快速比对。BWA 对于短读段的比对效率很高,并且提供了多种比对模式,包括 BWA-backtrack、BWA-SW 和 BWA-MEM。
- Bowtie 和 Bowtie2 是另外两个流行的对齐工具,它们利用了类似于 BWA 的算法。Bowtie 适用于较短的读段,而 Bowtie2 专门为更长的读段进行了优化。
- STAR 是一个 RNA-seq 数据对齐工具,它在处理长读段时表现出色。STAR 使用了“双阶段超快全面的RNA序列比对器”的策略,能够高效地处理大量数据。
这些工具在设计时考虑了不同类型的测序数据和不同的应用需求。用户需要根据实验设计选择最适合的对齐工具。
4.2 对齐策略的优化与评估
4.2.1 参数调整对结果的影响
在使用上述对齐工具时,必须仔细调整各种参数以达到最佳比对效果。对齐工具的参数调整对最终的比对质量有着显著的影响。例如,一个关键的参数是比对的错配容忍度(mismatch allowance)。如果错配容忍度设置得太低,一些由于测序误差导致的正常变异可能被错误地排除;而如果设置得太高,则可能导致不相关的读段被错误地比对到参考基因组上。
此外,比对算法选择和种子长度(seed length)也是关键参数。短种子长度可能降低比对的准确性,但有助于检测到更长的插入或缺失,而长种子长度能提供更高的准确性,但可能在存在较大变异的情况下失败。
4.2.2 对齐率和覆盖率的分析
对齐率是指有多少比例的测序读段成功地比对到了参考基因组上,而覆盖率则描述了参考基因组被测序读段覆盖的完整性。分析对齐率和覆盖率可以帮助我们评估测序实验的效率和质量。
一个高对齐率通常表示实验过程的质量较高,测序读段的质量较好。而覆盖率则告诉我们整个基因组有多少区域被有效覆盖,这对于后续的分析步骤至关重要。例如,在 CHIP-Seq 实验中,高覆盖率意味着我们能更准确地定位蛋白-DNA相互作用的位点。
4.2.3 整合多个样本数据的策略
在进行多个样本的测序实验时,整合样本数据可以增加统计能力并提高分析的可靠性。整合对齐数据的策略通常包括以下步骤:
- 归一化 :将不同样本的读段数量标准化,以消除样本间的差异。
- 整合 :使用如 BEDtools、GATK 或 SAMtools 等工具来合并多个样本的比对结果。
- 变体检测 :通过比较多个样本,识别基因组中的变异。
整合多个样本数据的关键在于确保所有样本都经过相同处理步骤的校准,并且使用相同或兼容的工具和参数进行对齐和后续分析。
4.3 对齐后数据的处理与检查
4.3.1 索引构建与数据检索
对齐后的数据需要构建索引以便于快速检索。索引是一种数据结构,它允许快速查找数据项而不需要查看整个数据集。例如,SAM/BAM 格式的文件使用索引文件(.bai),可以加速读取数据和随机访问。
构建索引可以使用 SAMtools 等工具。构建完成后,可以使用类似 samtools view 命令快速访问特定区域的数据。索引的构建对于后续的峰值检测、变异分析以及可视化展示至关重要。
4.3.2 对齐数据的可视化检查工具
数据可视化检查工具如 IGV(Integrative Genomics Viewer)允许研究人员对对齐后的数据进行直观的检查。使用 IGV,可以加载 BAM 文件并可视化特定基因组区域的读段分布。通过颜色编码读段的质量、方向和成对性,可以直观地识别潜在的问题区域。
4.3.3 假阳性与假阴性的识别
识别假阳性与假阴性是数据比对后分析的一个重要方面。假阳性指的是错误地认为某些读段与参考基因组匹配,而实际上它们可能来自于测序错误或样本污染。假阴性则指未能正确识别匹配的读段,这可能是由于测序深度不足或对齐算法对某些特殊区域处理不够好。
识别这些错误通常需要对数据进行质量控制、使用统计分析方法以及结合生物学背景知识。例如,利用 IGV 可视化检查读段分布,查看异常区域,并通过进一步的实验验证可疑读段的真实性。
代码块展示与逻辑分析
以下是一个使用 SAMtools 查看 BAM 文件内容的简单示例:
# 查看 BAM 文件头信息
samtools view -H example.bam
# 查看基因组特定区域的读段信息
samtools view example.bam chr1:10000-11000
# 使用 IGV 可视化查看 BAM 文件数据
igvtools toTDF example.bam example.tdf
igv & # 启动 IGV 图形界面
在上述示例中, -H 参数用于输出 BAM 文件的头部信息,这些信息包括文件的格式、使用的参考基因组等。接下来的命令展示了如何查看某个特定基因组区域的读段信息。最后, igvtools 命令用于将 BAM 文件转换为 IGV 可读的 TDF 文件格式,以便在 IGV 中可视化数据。
通过这些步骤,研究者可以检查数据的质量,验证数据的对齐情况,并准备数据用于进一步分析。这些操作不仅提供了数据可靠性的直接证据,还能帮助优化后续分析的参数设置。
5. 峰值检测技术与工具
5.1 峰值检测的理论基础
5.1.1 峰值定义与特征
峰值检测是指在CHIP-Seq数据分析过程中,识别出基因组上具有显著信号强度的区域。这些区域通常与转录因子结合位点或染色质修饰相关联。理论上来讲,一个峰值通常表现为信号强度的局部极大值,这些信号强度是由读段数(Read Count)或读段密度来衡量的。
5.1.2 峰值检测的统计模型
在峰值检测过程中,统计模型发挥着核心作用。最常用的统计模型包括泊松分布和负二项分布。泊松模型适用于背景噪音均匀,而信号较为集中,且没有超变异(Overdispersion)的情况。然而,CHIP-Seq数据往往存在超变异现象,此时负二项分布模型则更为合适。该模型能够处理额外的变异,更为精确地描述读段数的波动。
5.2 峰值检测工具的选择与应用
5.2.1 常用峰值检测软件介绍
目前市场上存在多种峰值检测软件,各有其特点。例如,MACS(Model-based Analysis of ChIP-Seq)是一个广泛使用的峰值检测工具,它利用滑动窗口(Sliding Window)来识别信号强度的局部极大值,并使用泊松模型或负二项分布模型进行统计检验。
另一个流行的工具是HOMER,它提供了多种峰值识别方法,包括二次模型分析。HOMER还能够处理大型数据集,并提供多种后续分析功能,如峰值注释和基序发现(Motif Finding)。
5.2.2 参数设置与调优技巧
不同的峰值检测软件具有不同的参数设置,优化这些参数对于获得高质量的峰值集至关重要。以下是一些普遍适用的参数调优技巧:
- 窗口大小(Window size) : 窗口大小会影响峰值的检测灵敏度和特异性。太大的窗口可能导致灵敏度降低,而太小的窗口可能引起假阳性增加。
- p值阈值(p-value threshold) : 该阈值用于判定一个峰值是否具有统计学意义。降低p值可以减少假阳性,但有可能遗漏一些重要的峰值。
- 信号与背景比(Signal to Noise Ratio, SNR) : 该参数用于区分有效信号与背景噪音。SNR设置较高会提高峰值识别的准确性。
5.2.3 案例分析:不同工具的性能对比
不同峰值检测工具的性能受多种因素影响,包括数据的复杂性、质量控制的严格程度以及峰值的生物学特征。通过实际案例分析,我们可以更直观地了解不同工具的优劣。例如,在一项研究中,将MACS与HOMER应用于同一组CHIP-Seq数据,并比较了它们识别出的峰值集的重叠度、信号强度、以及与生物学预期的吻合度。
通过表格列举两种工具的性能对比,可以帮助研究者做出更合理的工具选择:
| 工具 | 重叠度 | 信号强度 | 生物学吻合度 | |:----:|:------:|:--------:|:-------------:| | MACS | 高 | 较强 | 高 | | HOMER| 低 | 较弱 | 中 |
请注意,上表中的数据仅为示例,并不代表真实情况。
5.3 峰值注释与生物学意义关联
5.3.1 峰值区域的功能注释
峰值的生物功能注释是理解基因调控网络的关键步骤。通常,峰值被注释为可能的转录因子结合位点、增强子或其他染色质特征。这一过程通常涉及峰值与已知的基因组区域(如注释的基因、增强子、CpG岛等)的重叠分析。
5.3.2 转录因子结合位点分析
转录因子结合位点(Transcription Factor Binding Sites, TFBSs)分析是峰值注释的一个重要方面。通过对峰值区域的基序(Motif)分析,可以发现可能的转录因子结合位点,从而推断哪些转录因子可能参与了基因的调控。
5.3.3 峰值数据与基因表达的相关性分析
峰值数据与基因表达数据之间的相关性分析可以揭示峰值区域的生物学意义。通过比较峰值区域与基因表达水平的相关性,可以识别出可能的调控作用。例如,某基因的表达水平可能与其上游增强子区域的峰值信号强度正相关。
通过对 CHIP-Seq 数据中峰值的检测和深入分析,研究者可以更加精确地解析基因调控机制,为未来的生物医学研究提供重要基础。在下一章节中,我们将继续探讨如何对检测出的峰值进行深入分析和解释。
简介:CHIP-Seq技术是研究蛋白质-DNA相互作用的重要工具,尤其在理解基因表达调控方面发挥了巨大作用。本文介绍从实验准备到生物信息学分析的 CHIP-Seq 数据分析流程,包括质量控制、数据预处理、读取对齐、峰值检测、峰注释、差异分析、功能富集、网络构建以及结果可视化等关键步骤。

火山引擎开发者社区是火山引擎打造的AI技术生态平台,聚焦Agent与大模型开发,提供豆包系列模型(图像/视频/视觉)、智能分析与会话工具,并配套评测集、动手实验室及行业案例库。社区通过技术沙龙、挑战赛等活动促进开发者成长,新用户可领50万Tokens权益,助力构建智能应用。
更多推荐
 28
28 0
0- 0



 已为社区贡献16条内容
已为社区贡献16条内容

所有评论(0)