同时调用多种单细胞基础模型?!这个工具务必要用到自己的课题中(BioLLM)
单细胞 RNA 测序(scRNA-seq)通过实现高分辨率转录组分析,彻底改变了传统分子生物学。已经开发了几种基础模型来分析大规模的单细胞测序数据,如 scBERT, Geneformer, scGPT 和 scFoundation。然而,这些模型不仅在架构设计和预训练策略上表现出一定差异,而且数据集大小和参数数量也有所不同。
生信碱移
通用单细胞基础模型
BioLLM 提供了通用的调用方案,将各种单细胞基础模型无缝连接到一个生态系统中。
单细胞 RNA 测序(scRNA-seq)通过实现高分辨率转录组分析,彻底改变了传统分子生物学。已经开发了几种基础模型来分析大规模的单细胞测序数据,如 scBERT, Geneformer, scGPT 和 scFoundation。然而,这些模型不仅在架构设计和预训练策略上表现出一定差异,而且数据集大小和参数数量也有所不同。例如,scBERT 采用通过掩码语言建模训练的双向 transformer,并结合 gene2vec 嵌入来学习基因表示 ,而 scGPT 则采用自回归训练策略,使用 Flash-Attention 块和随机基因嵌入,专注于为每个细胞生成嵌入。鉴于上述差异,每个模型在各种下游任务(如批次效应校正和细胞类型分类)中的表现可能会有显著不同,所以应当针对不同任务选择合适的模型。
虽然一些模型,如 Geneformer 和 scGPT,提供了详尽的文档和结构良好的开源代码库,便于研究人员应用和定制自己的分析内容,但大部分模型可能缺乏全面的教学文档以及统一的代码运行过程。为此,来自中国科学院大学与华大基因的研究者整合开发了一款工具 BioLLM。BioLLM 提供了通用的调用方案,将各种单细胞基础模型无缝连接到一个生态系统中。借助标准化的 API 和全面的文档,BioLLM 简化了各种单细胞基础模型之间的切换和比较分析。
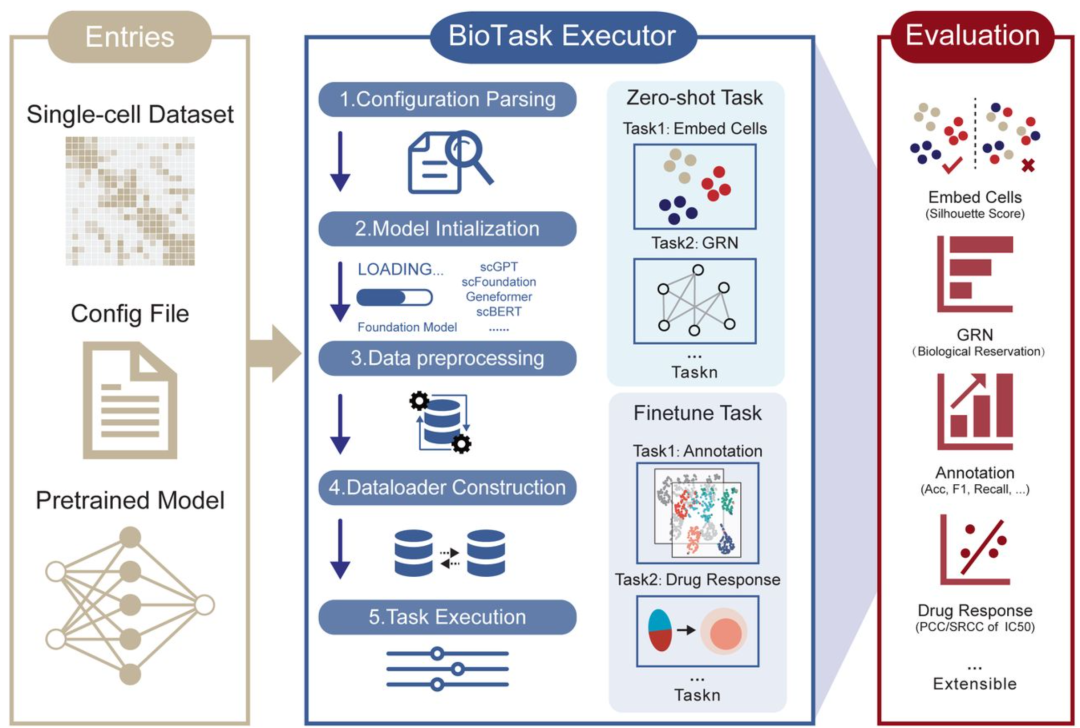
▲ BioLLM 单细胞数据分析框架:BioLLM 框架由Entries、BioTask Executor 和 Evaluation 三个组件组成。Entries 包括输入数据集、配置文件和预训练模型;BioTask Executor 通过五个步骤处理任务:Configuration Parsing、Model Initialization、Data Preprocessing、Dataloader Construction 和 Task Execution(包括 zero-shot 和 fine-tuning 任务);Evaluation 涉及细胞嵌入(平均轮廓宽度,ASW)、GRN 分析(基因本体富集)、细胞类型注释(准确率、精确率、召回率、宏 F1)以及药物反应预测(PCC、SRCC)。DOI: 10.1101/2024.11.22.624786。
除此之外,作者还对现有的主流单细胞基础模型进行了基准测试,展示了不同单细胞基础模型各自的优势和局限性。他们发现,scGPT 在所有任务中具有最强劲的表现。Geneformer 和 scFoundation 在基因级任务中展示了强大的能力,可能得益于有效的预训练策略。 相比之下,scBERT 的表现较差,这可能是由于其参数数量较少以及训练数据集的规模有限。
该工具的github链接如下,大家可以自行学习相关的文档教程。本文简要介绍如何基于 BioLLM 进行细胞嵌入的任务:
-
https://github.com/BGIResearch/BioLLM
-
https://github.com/BGIResearch/BioLLM/tree/master/tutorials
▲ BioLLM 支持使用单细胞基础模型进行多种下游任务,并附有相关文档。
0.BioLLM安装
可以使用以下代码进行安装:
git clone https://github.com/BGIResearch/BioLLM.git
cd BioLLM
python ./setup.py
需要注意的是,scGPT所需的flash-attn依赖库需要特定版本的 GPU 和 CUDA 驱动,作者建议使用 CUDA 11.7 和 flash-attn<1.0.5。
1.简单示例
BioLLM 将各种单细胞基础模型无缝连接到一个 python 生态库中,可以对接 scanpy 的下游分析。下面给各位佬哥展示一下如何基于 BioLLM 使用 scGPT 的模型进行细胞嵌入表示,可以用于细胞注释、批次矫正、嵌入空间分析等任务:
① 读入数据,共计 8978 个细胞,36398 个基因:
import scanpy as sc # scanpy单细胞分析生态
# adata = sc.read_h5ad("data/trajectory/trajectory_seurat_filtered.h5ad")
adata = sc.read_h5ad("/home/share/huadjyin/home/s_qiuping1/workspace/omics_model/bio_model/biollm/case/data/zero-shot/bone_marrow/bone_marrow.h5ad")
adata
#AnnData object with n_obs × n_vars = 8978 × 36398
# obs: 'cell.labels', 'lanes', 'assay_ontology_term_id', 'cell_type_ontology_term_id', 'development_stage_ontology_term_id', 'disease_ontology_term_id', 'donor_id', 'is_primary_data', #'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'sex_ontology_term_id', 'suspension_type', 'tissue_ontology_term_id', 'tissue_type', 'cell_type', 'assay', 'dise#ase', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
# var: 'feature_types', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'gene_symbols', 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length'
# uns: 'antibody.X', 'antibody_features', 'antibody_raw.X', 'citation', 'default_embedding', 'rank_genes_groups', 'rank_genes_groups_filtered', 'schema_reference', 'schema_version', 'title'
# obsm: 'X_umap'
② 常规流程,标准化>>高变基因>>线性降维>>非线性降维。其实示例数据已经做了这些分析,为了展示还是把流程贴上来,这里可以展示细胞注释降维图:
# 标准化
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 高变基因
adata.var_names = adata.var['feature_name']
sc.pp.highly_variable_genes(adata, n_top_genes=2000, subset=True)
# 线性降维
sc.tl.pca(adata)
# 非线性降维
sc.pp.neighbors(adata, n_neighbors=4, n_pcs=20)
sc.tl.umap(adata, min_dist=0.6, spread=1.5)
# 展示预先定义的细胞类型cell_type
sc.pl.umap(adata, color = ['cell_type'])

③ 基于scGPT的细胞嵌入结果。简单理解,基础模型见过更多的数据(具有一定先验),对细胞的降维结果会比原本的PCA要更好,这里只是用 scGPT 的嵌入代替的 pca 做下游分析(其实也是各种单细胞模型以及空转领域的常见做法):
from biollm.utils.utils import load_config
from biollm.base.load_scgpt import LoadScgpt
import os
import scanpy as sc
# 模型配置
config_file = './configs/scgpt_cell_emb.toml'
configs = load_config(config_file)
# get_embedding 函数获取细胞嵌入
obj = LoadScgpt(configs)
file_name = os.path.basename(configs.input_file)
obj.model = obj.model.to(configs.device)
emb = obj.get_embedding(obj.args.emb_type, adata=adata)
adata.obsm['scgpt_cell_emb'] = emb
print('embedding shape:', emb.shape)
#{'ntoken': 60697, 'd_model': 512, 'nhead': 8, 'd_hid': 512, 'nlayers': 12, 'nlayers_cls': 3, 'n_cls': 1, 'dropout': 0.2, 'pad_token': '<pad>', 'do_mvc': False, 'do_dab': False, 'use_batch_lab#els': False, 'num_batch_labels': None, 'domain_spec_batchnorm': False, 'input_emb_style': 'continuous', 'cell_emb_style': 'cls', 'mvc_decoder_style': 'inner product', 'ecs_threshold': #0.3, 'explicit_zero_prob': False, 'fast_transformer_backend': 'flash', 'pre_norm': False, 'vocab': GeneVocab(), 'pad_value': -2, 'n_input_bins': 51, 'use_fast_transformer': True}
重新做个非线性降维试试,现在使用的是 scGPT 的嵌入 scgpt_cell_emb:
sc.pp.neighbors(adata, use_rep='scgpt_cell_emb', n_neighbors=5, n_pcs = 30)
sc.tl.umap(adata)
sc.pl.umap(adata, color='cell_type')
sc.tl.draw_graph(adata)

会不会更加具有区分度呢
见仁见智吧
基因敲除啥的任务大家也可以试试
欢迎各位佬哥佬姐关注

火山引擎开发者社区是火山引擎打造的AI技术生态平台,聚焦Agent与大模型开发,提供豆包系列模型(图像/视频/视觉)、智能分析与会话工具,并配套评测集、动手实验室及行业案例库。社区通过技术沙龙、挑战赛等活动促进开发者成长,新用户可领50万Tokens权益,助力构建智能应用。
更多推荐
 25
25 0
0- 0



 已为社区贡献6条内容
已为社区贡献6条内容

所有评论(0)